Laboratorio Biotecnologie
Estrazione
di DNA da matrici alimentari vegetali e allestimenti di reazioni di PCR
(reazione a catena della polimerasi) con primer specifici per il DNA vegetale e
per il DNA batterico.

Obiettivi:
● Purificare il DNA da cellule vegetali per la tipizzazione della varietà. Le varietà analizzate sono state: caffè, cacao, curcuma, foglie di banano.
● Purificare il DNA batterico se presente e identificare la specie prevalente.
Attività pratiche effettuate in laboratorio
● Estrazione di DNA con kit commerciale.
● Elettroforesi su gel di agarosio.
● PCR per DNA vegetale e batterico.
Fasi estrazione del DNA:
● Distruzione della parete cellulare.
● Lisi cellulare e precipitazioni di proteine, carboidrati e lipidi.
● Rimozione dell’ RNA.
● Precipitazione e purificazione del DNA.
Preparazione:
● Mischiare completamente i reagenti.
● Esaminare i reagenti per precipitazione.
● Se uno dei reagenti forma un precipitato, riscaldare a 55/65° C fino a che il precipitato si dissolve e permette di essere stabilizzato a temperatura ambiente prima dell’uso.
● Pre-riscaldare il bagnetto d'acqua a 65°C.
● Diluire la soluzione di lavaggio concentrata con 9.5ml, 72 ml, 330 ml di etanolo al 95-100%.
● Pre-riscaldare la soluzione di eluizione a 65°C.
Procedura:
1. Distruggere le cellule: macinare il tessuto delle piante in una polvere fine nel liquido di idrogeno usando un mortaio. Trasferire fino a 100 mg della polvere in un tubo di microcentrifuga.Tenere il campione nel ghiaccio o congelarlo a -70°C.
2. Lisi delle cellule: aggiungere 350 microL di soluzione di lisi ( parte a ) 50 microL di soluzione di lisi ( parte b ) al tubo; mischiare completamente . Un precipitato bianco si formerà sopra l'aggiunta della soluzione di lisi parte b. Incubare il miscuglio a 65 °C per 10 minuti con inversioni occasionali per dissolvere il precipitato. Digestione opzionale con RNasi: questo kit è disegnato per isolare in modo selettivo grandi parti di DNA . Se le preparazioni si rivelano essere contaminate con RNA , RNasi può essere usato per digerire RNA. Aggiungere 50 unità di RNasi al miscuglio di lisi appena prima dell' incubazione a 65°C.
3. Precipitazione dei detriti : aggiungere 130 microL di soluzione di precipitazione al miscuglio; mischiare completamente per inversione e porre il campione nel ghiaccio per 5 minuti; centrifugare il campione a velocità massima per 5 minuti per dividere i detriti cellulari, le proteine e i polisaccaridi.
4. Filtrazione dei detriti: pipettare attentamente il surnatante dallo step tre nel GenElute colonna di filtrazione centrifugare alla massima velocità per 1 minuto, questo rimuove qualsiasi detrito cellulare non rimosso nello step tre, rimuovere la colonna di filtrazione, ma tenere il tubo di collezione, preparare per il legame, aggiungere 70 microl di soluzione di legame direttamente al liquido dello step 4, mischiare completamente per inversione.
5. Preparare per il legame: aggiungere 700 microL di soluzione di legame direttamente al liquido ottenuto dallo step 4. Mischiare a fondo tramite inversione.
6. Preparare la colonna di legame: inserire un colonna di legame miniprep GenElute (con un anello rosso) in un tubo fornito per la microcentrifuga, se non già assemblato. Aggiungere 500 microL della soluzione di preparazione della colonna a ciascun miniprep e centrifugare a 12000 Xg da 30 secondi a un minuto. Scartare il liquido ottenuto.
Nota: La colonna di preparazione della soluzione massimizza il legame del DNA alla membrana.
7. Caricare il lisato: pipettare con cura 700 microL della miscela dello step 5 sulla colonna preparata nello step 6 e centrifugare a massima velocità per un minuto. Scartare il liquido ottenuto; mantenere il tubo di raccolta. Riportare la colonna nel tubo di raccolta. Inserire il lisato rimanente dello step 5 nella colonna. Ripetere la centrifuga come sopra e scartare il liquido ottenuto e il tubo di raccolta.
8. Primo lavaggio della colonna: prima dell'uso assicurarsi di aggiungere etanolo alla soluzione concentrata di lavaggio. Porre la colonna di legame in un tubo collettore fresco di 2 mL e aggiungere 500 microL della soluzione di lavaggio diluita alla colonna. Centrifugare alla velocità massima per un minuto. Eliminare il liquido ma mantenere il tubo collettore.
9. Seconda colonna di lavaggio: aggiungere alla colonna altri 500 microL di soluzione di lavaggio diluita e centrifugare alla massima velocità per 3 minuti per asciugare la colonna. Non permettere al liquido ottenuto di toccare la colonna; pulire ogni fluido che è fuoriuscito.
10. Diluire il DNA. Trasferire la colonna di legame a 2mL da un tubo collettore nuovo. Applicare 100 microL di soluzione di eluizione preriscaldata (65° C) alla colonna e centrifugare alla massima velocità per un minuto. Ripetere l 'eluizione. Non permettere che il liquido venga a contatto con la colonna. Gli eluiti possono essere raccolti nello stesso tubo collettore. In alternativa, un secondo tubo di collezione può essere utilizzato un secondo tubo collettore per la seconda eluizione in modo da prevenire la diluizione del primo eluito.
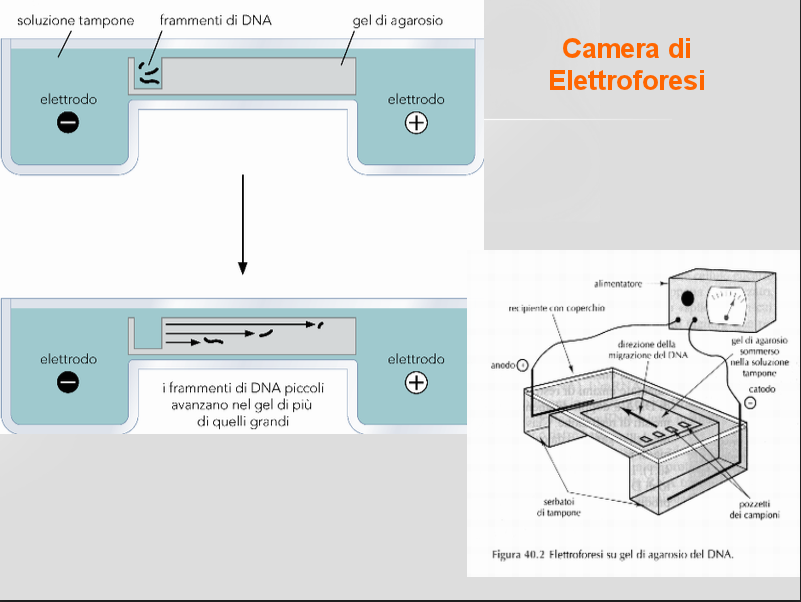
Elettroforesi su gel di agarosio
Questa tecnica ci consente di separare i frammenti di DNA purificati in base alle loro dimensioni.
Tecnica che utilizza le differenze di carica elettrica per separare le molecole presenti in una miscela. Le molecole di DNA sono cariche negativamente quindi se poste in un campo elettrico migrano verso il polo positivo.
● Purificare il DNA da cellule vegetali per la tipizzazione della varietà. Le varietà analizzate sono state: caffè, cacao, curcuma, foglie di banano.
● Purificare il DNA batterico se presente e identificare la specie prevalente.
Attività pratiche effettuate in laboratorio
● Estrazione di DNA con kit commerciale.
● Elettroforesi su gel di agarosio.
● PCR per DNA vegetale e batterico.
Fasi estrazione del DNA:
● Distruzione della parete cellulare.
● Lisi cellulare e precipitazioni di proteine, carboidrati e lipidi.
● Rimozione dell’ RNA.
● Precipitazione e purificazione del DNA.
Preparazione:
● Mischiare completamente i reagenti.
● Esaminare i reagenti per precipitazione.
● Se uno dei reagenti forma un precipitato, riscaldare a 55/65° C fino a che il precipitato si dissolve e permette di essere stabilizzato a temperatura ambiente prima dell’uso.
● Pre-riscaldare il bagnetto d'acqua a 65°C.
● Diluire la soluzione di lavaggio concentrata con 9.5ml, 72 ml, 330 ml di etanolo al 95-100%.
● Pre-riscaldare la soluzione di eluizione a 65°C.
Procedura:
1. Distruggere le cellule: macinare il tessuto delle piante in una polvere fine nel liquido di idrogeno usando un mortaio. Trasferire fino a 100 mg della polvere in un tubo di microcentrifuga.Tenere il campione nel ghiaccio o congelarlo a -70°C.
2. Lisi delle cellule: aggiungere 350 microL di soluzione di lisi ( parte a ) 50 microL di soluzione di lisi ( parte b ) al tubo; mischiare completamente . Un precipitato bianco si formerà sopra l'aggiunta della soluzione di lisi parte b. Incubare il miscuglio a 65 °C per 10 minuti con inversioni occasionali per dissolvere il precipitato. Digestione opzionale con RNasi: questo kit è disegnato per isolare in modo selettivo grandi parti di DNA . Se le preparazioni si rivelano essere contaminate con RNA , RNasi può essere usato per digerire RNA. Aggiungere 50 unità di RNasi al miscuglio di lisi appena prima dell' incubazione a 65°C.
3. Precipitazione dei detriti : aggiungere 130 microL di soluzione di precipitazione al miscuglio; mischiare completamente per inversione e porre il campione nel ghiaccio per 5 minuti; centrifugare il campione a velocità massima per 5 minuti per dividere i detriti cellulari, le proteine e i polisaccaridi.
4. Filtrazione dei detriti: pipettare attentamente il surnatante dallo step tre nel GenElute colonna di filtrazione centrifugare alla massima velocità per 1 minuto, questo rimuove qualsiasi detrito cellulare non rimosso nello step tre, rimuovere la colonna di filtrazione, ma tenere il tubo di collezione, preparare per il legame, aggiungere 70 microl di soluzione di legame direttamente al liquido dello step 4, mischiare completamente per inversione.
5. Preparare per il legame: aggiungere 700 microL di soluzione di legame direttamente al liquido ottenuto dallo step 4. Mischiare a fondo tramite inversione.
6. Preparare la colonna di legame: inserire un colonna di legame miniprep GenElute (con un anello rosso) in un tubo fornito per la microcentrifuga, se non già assemblato. Aggiungere 500 microL della soluzione di preparazione della colonna a ciascun miniprep e centrifugare a 12000 Xg da 30 secondi a un minuto. Scartare il liquido ottenuto.
Nota: La colonna di preparazione della soluzione massimizza il legame del DNA alla membrana.
7. Caricare il lisato: pipettare con cura 700 microL della miscela dello step 5 sulla colonna preparata nello step 6 e centrifugare a massima velocità per un minuto. Scartare il liquido ottenuto; mantenere il tubo di raccolta. Riportare la colonna nel tubo di raccolta. Inserire il lisato rimanente dello step 5 nella colonna. Ripetere la centrifuga come sopra e scartare il liquido ottenuto e il tubo di raccolta.
8. Primo lavaggio della colonna: prima dell'uso assicurarsi di aggiungere etanolo alla soluzione concentrata di lavaggio. Porre la colonna di legame in un tubo collettore fresco di 2 mL e aggiungere 500 microL della soluzione di lavaggio diluita alla colonna. Centrifugare alla velocità massima per un minuto. Eliminare il liquido ma mantenere il tubo collettore.
9. Seconda colonna di lavaggio: aggiungere alla colonna altri 500 microL di soluzione di lavaggio diluita e centrifugare alla massima velocità per 3 minuti per asciugare la colonna. Non permettere al liquido ottenuto di toccare la colonna; pulire ogni fluido che è fuoriuscito.
10. Diluire il DNA. Trasferire la colonna di legame a 2mL da un tubo collettore nuovo. Applicare 100 microL di soluzione di eluizione preriscaldata (65° C) alla colonna e centrifugare alla massima velocità per un minuto. Ripetere l 'eluizione. Non permettere che il liquido venga a contatto con la colonna. Gli eluiti possono essere raccolti nello stesso tubo collettore. In alternativa, un secondo tubo di collezione può essere utilizzato un secondo tubo collettore per la seconda eluizione in modo da prevenire la diluizione del primo eluito.
Elettroforesi su gel di agarosio
Questa tecnica ci consente di separare i frammenti di DNA purificati in base alle loro dimensioni.
Tecnica che utilizza le differenze di carica elettrica per separare le molecole presenti in una miscela. Le molecole di DNA sono cariche negativamente quindi se poste in un campo elettrico migrano verso il polo positivo.
|
|
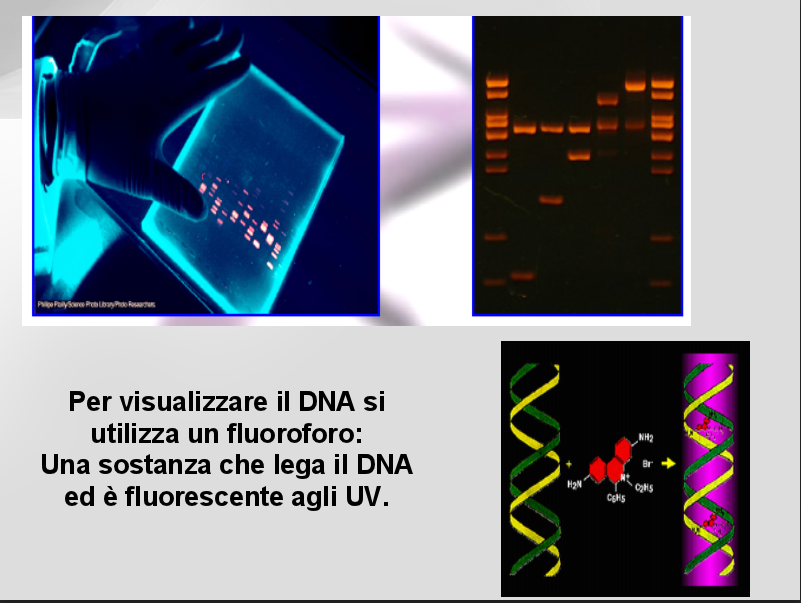
Per visualizzare il DNA si utilizza un fluoroforo.
PCR Polymerase Chain Reaction
E’ la tecnica utilizzata per la sintesi ripetitiva di specifiche regioni di DNA mediante l’uso dell’ enzima DNA polimerasi (Taq polimerasi ).
Componenti della reazione:
- DNA
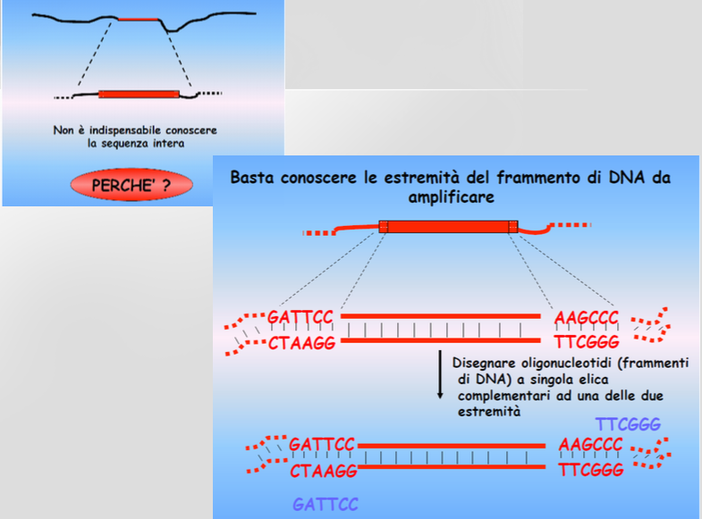
- Primers che fungono da innesco per i geni che voglio amplificare: per le piante geni cloroplastici come rbcl (Rubisco ); per i batteri vengono generalmente utilizzate le regioni variabili dei 16S. Sono composti da brevi sequenze nucleotidiche.
- Taq polimerasi temostabile isolata da Thermus aquaticus.
- dNTP ( dATP; dTTP; dCTP; dGTP)
- MgCl2
- pH buffer
E’ la tecnica utilizzata per la sintesi ripetitiva di specifiche regioni di DNA mediante l’uso dell’ enzima DNA polimerasi (Taq polimerasi ).
Componenti della reazione:
- DNA
- Primers che fungono da innesco per i geni che voglio amplificare: per le piante geni cloroplastici come rbcl (Rubisco ); per i batteri vengono generalmente utilizzate le regioni variabili dei 16S. Sono composti da brevi sequenze nucleotidiche.
- Taq polimerasi temostabile isolata da Thermus aquaticus.
- dNTP ( dATP; dTTP; dCTP; dGTP)
- MgCl2
- pH buffer
Reazione a Catena della Pcr
1) Denaturazione: la miscela di reazione viene riscaldata alla temperatura di circa 94°C. Ciò induce la denaturazione del DNA.
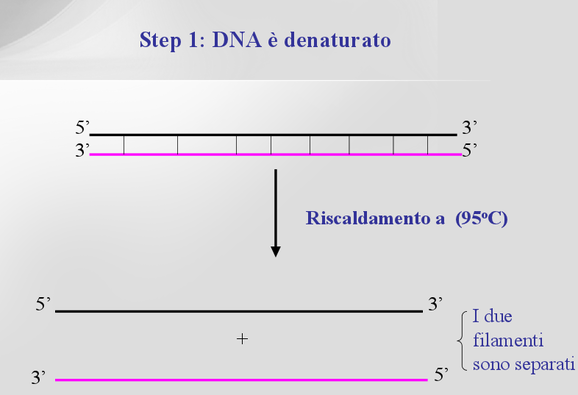
2) Appaiamento
o annealing: si abbassa la temperatura a valori di 58°C per permettere ai
primer di legarsi alle estremità dei due filamenti di DNA.
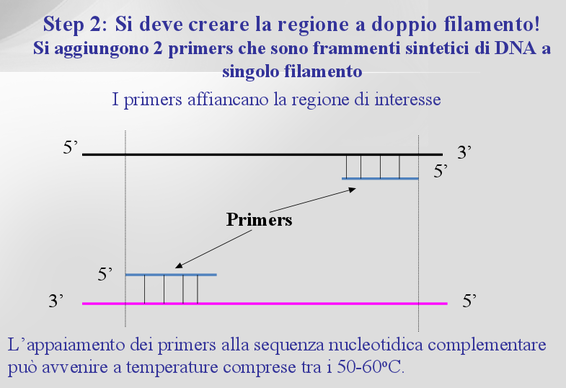
3) Estensione: la DNA polimerasi in presenza di Mg++, permette la sintesi di nuovi filamenti a partire dai due primer in direzione 5’-3’. La temperatura è di circa 72°C.
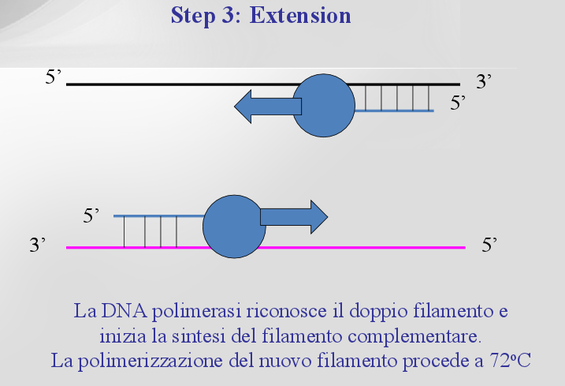
Questi tre passaggi sono poi ripetuti per n cicli ( in genere 25-35, che durano ciascuno pochi minuti), portando a un numero di frammenti identici pari a 2^n e avvengono in modo automatizzato grazie ai termociclatori, macchinari programmati per effettuare i vari cicli di riscaldamento e di raffreddamento.
Laboratorio di bioinformatica

Si tratta dell’utilizzo dei più comuni software disponibili online per le analisi delle sequenze di DNA.
In questa sezione viene illustrato come si procede all’analisi delle sequenze di DNA ottenute tramite i processi di laboratorio, per verificare le effettive caratteristiche del prodotto analizzato con delle tabelle di riscontro contenenti dati relativi a miliardi di sostanze.
Il procedimento che abbiamo seguito ė costituito dalle seguenti fasi:
• Scaricare Chromas, programma per la visualizzazione di grafici relativi alle sequenze di basi delle diverse sostanze. (Chromas, open, edit)
• Aprire la pagina web NCBI (National Centre for Biotechnology Information) http://www.ncbi.nlm.nih.gov
• Selezionare BLAST (Basic Local Alignement Search Tool) in Popular Resources.
• Selezionare nucleotide blast (sezione per la ricerca di sequenze nucleotidiche nei database immettendo la propria sequenza), nucleotide collection e per ultimo mega blast.
• Dopo avere immesso la sequenza (query sequence), ricavata da Chromas, copiando la sequenza (edit > copy sequence > FASTA format) nell'apposito spazio, selezionare BLAST in fondo alla pagina.
In questa sezione viene illustrato come si procede all’analisi delle sequenze di DNA ottenute tramite i processi di laboratorio, per verificare le effettive caratteristiche del prodotto analizzato con delle tabelle di riscontro contenenti dati relativi a miliardi di sostanze.
Il procedimento che abbiamo seguito ė costituito dalle seguenti fasi:
• Scaricare Chromas, programma per la visualizzazione di grafici relativi alle sequenze di basi delle diverse sostanze. (Chromas, open, edit)
• Aprire la pagina web NCBI (National Centre for Biotechnology Information) http://www.ncbi.nlm.nih.gov
• Selezionare BLAST (Basic Local Alignement Search Tool) in Popular Resources.
• Selezionare nucleotide blast (sezione per la ricerca di sequenze nucleotidiche nei database immettendo la propria sequenza), nucleotide collection e per ultimo mega blast.
• Dopo avere immesso la sequenza (query sequence), ricavata da Chromas, copiando la sequenza (edit > copy sequence > FASTA format) nell'apposito spazio, selezionare BLAST in fondo alla pagina.
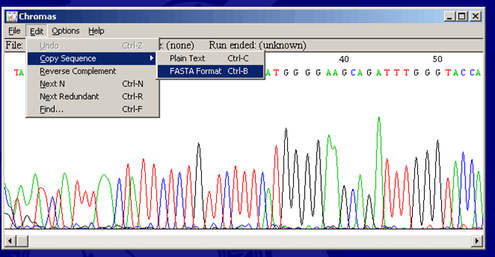
Interfaccia Chromas
• Si ottiene la BLAST TREE VIEW, che rappresenta un “albero genealogico” della nostra sostanza.
• Aprire la pagina web.expasy.org/translate/ per verificare quanto la sostanza analizzata corrisponde a una sostanza presente nel database, così da scoprire eventuali contraffazioni.
• Aprire Translate tool, incollare nella finestra la sequenza nucleotidica, per ottenere la sequenza amminoacidica; così si otterranno più possibilità per il frammento di coltura.
• identificare il frame corretto, ovvero quello privo di codoni di stop, e selezionare la metionina che apre la lettura, si ottiene così la sequenza polipeptidica della proteina.i
• Si ottiene la BLAST TREE VIEW, che rappresenta un “albero genealogico” della nostra sostanza.
• Aprire la pagina web.expasy.org/translate/ per verificare quanto la sostanza analizzata corrisponde a una sostanza presente nel database, così da scoprire eventuali contraffazioni.
• Aprire Translate tool, incollare nella finestra la sequenza nucleotidica, per ottenere la sequenza amminoacidica; così si otterranno più possibilità per il frammento di coltura.
• identificare il frame corretto, ovvero quello privo di codoni di stop, e selezionare la metionina che apre la lettura, si ottiene così la sequenza polipeptidica della proteina.i
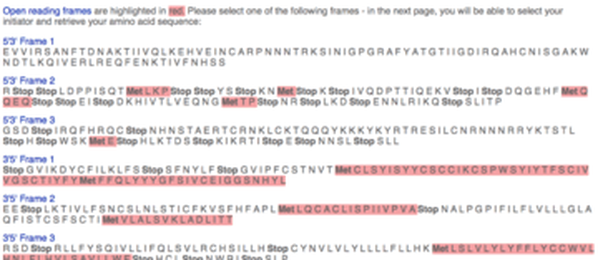

• Utilizzando di nuovo BLAST (protein Blast) e inserendo la sequenza polipeptidica sarà possibile
identificare la proteina e l'organismo a cui è associata (attraverso il Blast Tree View)
La biodiversità può essere misurata anche a livello morfologico.
A questo scopo risulta molto utile il sito UPOV- union for the protection of new varieties of plants.
Selezionando nel sito UPOV “Test Guidelines” e poi “Search Test Guidelines”, si ha la possibilità di scegliere tra varie specie di vegetali come ad esempio il ciliegio, quindi selezionando una di queste ne saranno visibili i descrittori morfologici (forma della foglia ecc.)
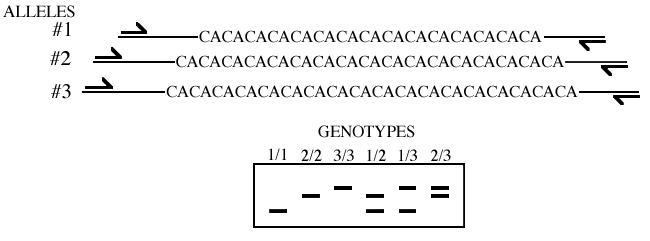
Oggi per la misurazione della biodiversità si usano marcatori molecolari che sono polimorfici tra gli individui di una popolazione (ad esempio i Microsatelliti).
Con i marcatori si cercano i primer a monte e a valle, il che consente di eseguire la PCR (reazione a catena della polimerasi).
La biodiversità può essere misurata anche a livello morfologico.
A questo scopo risulta molto utile il sito UPOV- union for the protection of new varieties of plants.
Selezionando nel sito UPOV “Test Guidelines” e poi “Search Test Guidelines”, si ha la possibilità di scegliere tra varie specie di vegetali come ad esempio il ciliegio, quindi selezionando una di queste ne saranno visibili i descrittori morfologici (forma della foglia ecc.)
Oggi per la misurazione della biodiversità si usano marcatori molecolari che sono polimorfici tra gli individui di una popolazione (ad esempio i Microsatelliti).
Con i marcatori si cercano i primer a monte e a valle, il che consente di eseguire la PCR (reazione a catena della polimerasi).